Abstract
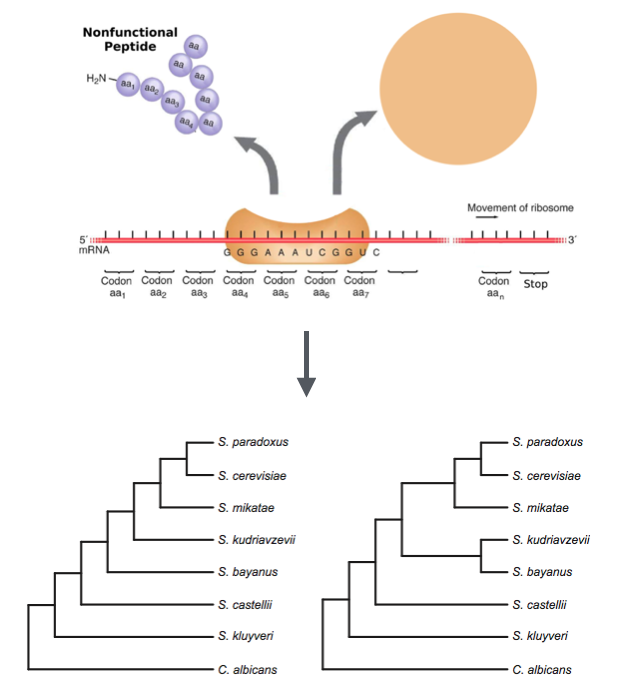
The quality of phylogenetic inference made from protein-coding genes depends, in part, on the realism with which the codon substitution process is modeled. Here we propose a new mechanistic model that combines the standard M0 substitution model of Yang (1997) with a simplified model from Gilchrist (2007) that includes selection on synonymous substitutions as a function of codon-specific nonsense error rates. We tested the newly proposed model by applying it to 104 protein-coding genes in brewer’s yeast, and compared the fit of the new model to the standard M0 model and to the mutation–selection model of Yang and Nielsen (2008) using the AIC. Our new model provided significantly better fit in approximately 85% of the cases considered for the basic M0 model and in approximately 25% of the cases for the M0 model with estimated codon frequencies, but only in a few cases when the mutation–selection model was considered. However, our model includes a parameter that can be interpreted as a measure of the rate of protein production, and the estimates of this parameter were highly correlated with an independent measure of protein production for the yeast genes considered here. Finally, we found that in some cases the new model led to the preference of a different phylogeny for a subset of the genes considered, indicating that substitution model choice may have an impact on the estimated phylogeny.