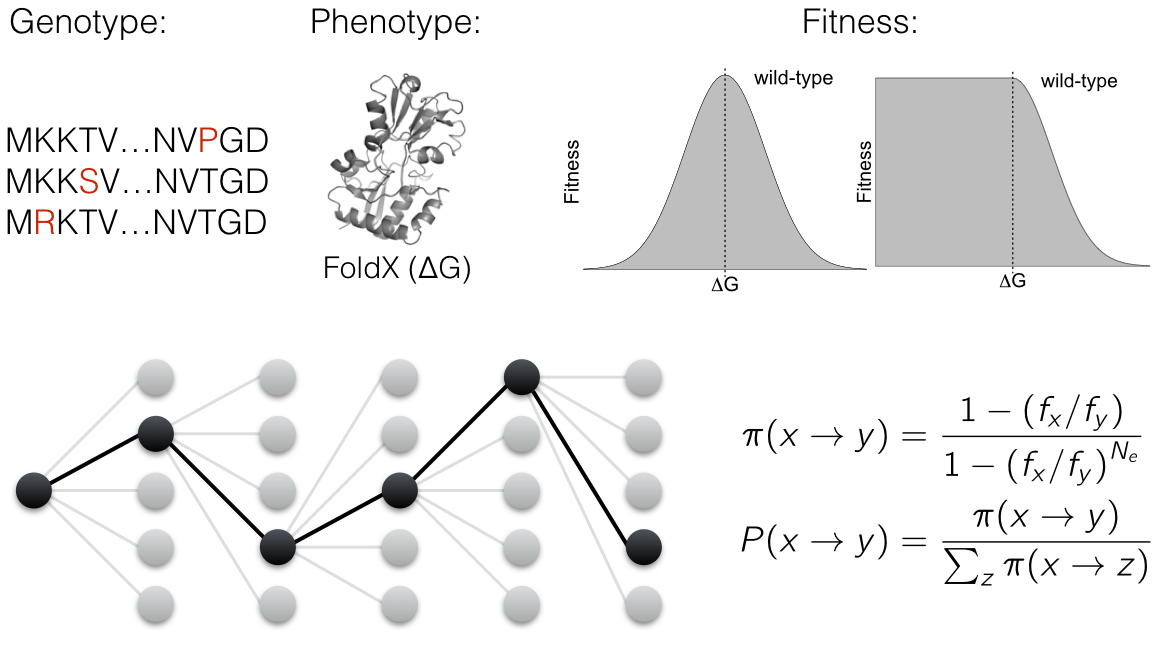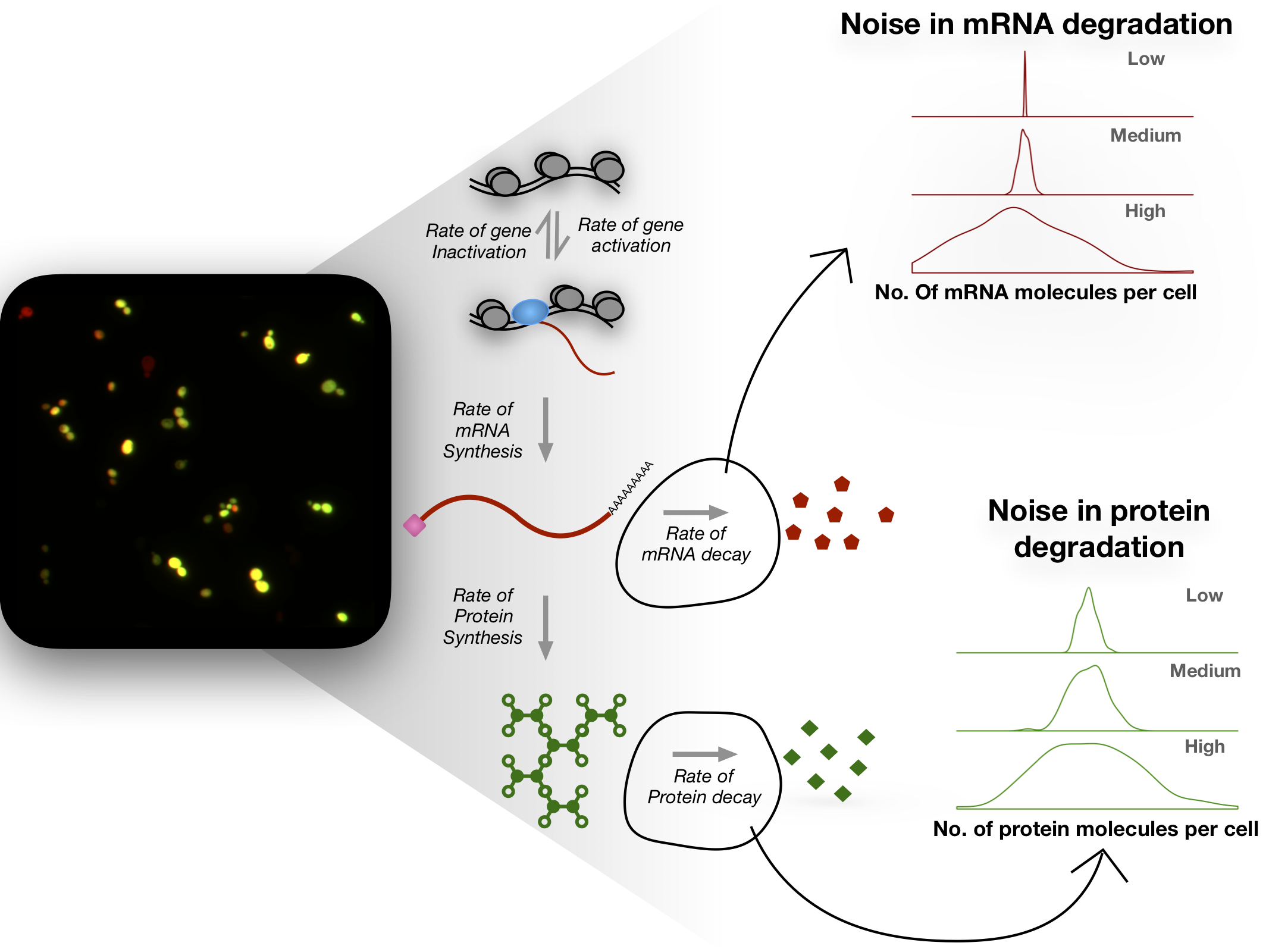
Gene expression is a highly stochastic process leading to variability in mRNA and protein abundances even within an isogenic population of cells grown in the same environment. This heterogeneity is commonly referred to as noise in gene expression. Biochemical processes such as transcription, translation, and mRNA and protein decay are each noisy and contribute to the overall variability in gene expression. While noise in bacteria is thought to be predominantly driven by translation, noise in eukaryotes has been attributed mainly to transcriptional processes. However, the stochastic nature of mRNA and protein decay has often been ignored when estimating noise. As a result, noise due to transcription or translation is likely overestimated.
Also, sequence features of an mRNA that contribute to noise remain unexplored. Using single-molecule FISH and flow cytometry (FISH-Flow), we are studying the variation in mRNA and protein decay in single cells. Understanding how decay dynamics of mRNAs and proteins contribute to noise will help model noise more accurately, and provide insights into how living systems regulate noise through various biochemical processes