Abstract
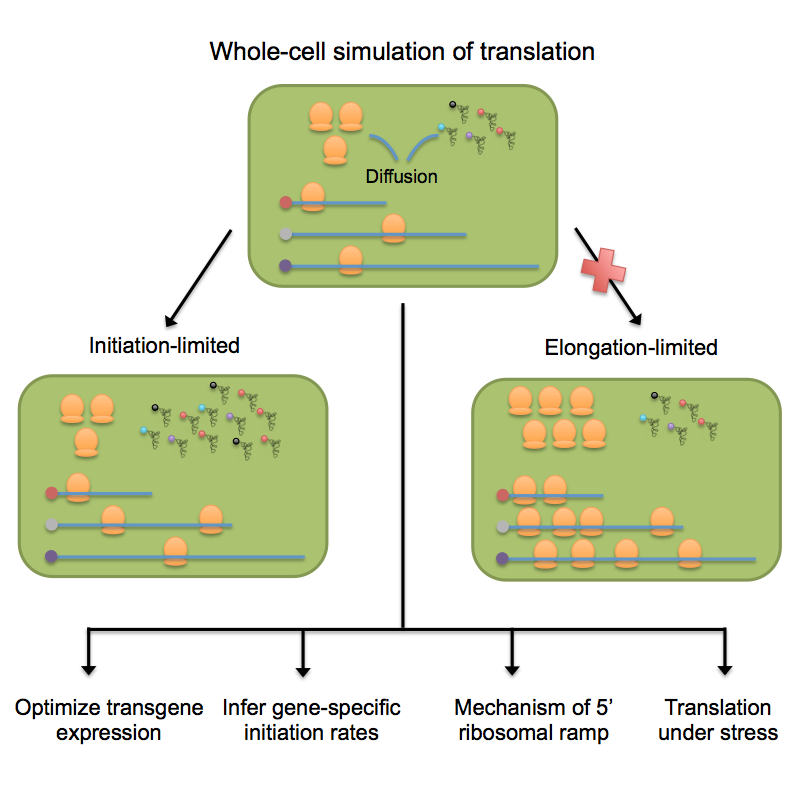
Deep sequencing now provides detailed snapshots of ribosome occupancy on mRNAs. We leverage these data to parameterize a computational model of translation, keeping track of every ribosome, tRNA, and mRNA molecule in a yeast cell. We determine the parameter regimes in which fast initiation or high codon bias in a transgene increases protein yield and infer the initiation rates of endogenous Saccharomyces cerevisiae genes, which vary by several orders of magnitude and correlate with 5′ mRNA folding energies. Our model recapitulates the previously reported 5′-to-3′ ramp of decreasing ribosome densities, although our analysis shows that this ramp is caused by rapid initiation of short genes rather than slow codons at the start of transcripts. We conclude that protein production in healthy yeast cells is typically limited by the availability of free ribosomes, whereas protein production under periods of stress can sometimes be rescued by reducing initiation or elongation rates.
Stochastic Model of Protein Translation
The associated code to simulate whole-cell model of protein translation is available on GitHub. The model keeps track of all ribosomes, tRNAs and mRNAs in a cell in continuous time. The rates of initiation and elongation in the model are based on physical parameters that have been determined experimentally in yeast.
The pacakge contains the source code, pre-compiled binaries, utility scripts and example input and output files. The source code is licensed under GNU GPLv3 license.
Commentary
Here's the associated commentary on our paper that was featured on Penn News.