Abstract
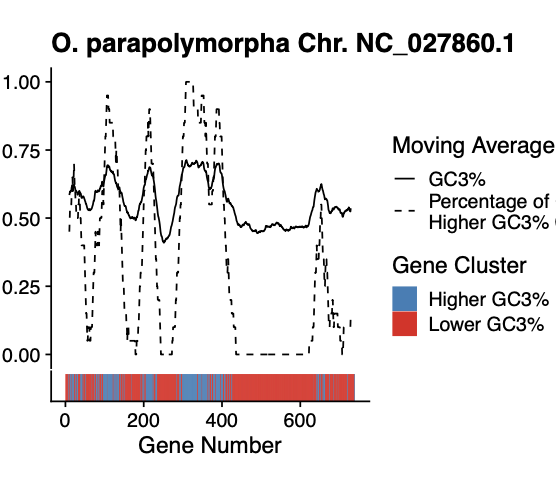
Patterns of non-uniform usage of synonymous codons (codon bias) varies across genes in an organism and across species from all domains of life. The bias in codon usage is due to a combination of both non-adaptive (e.g. mutation biases) and adaptive (e.g. natural selection for translation efficiency/accuracy) evolutionary forces. Most population genetics models quantify the effects of mutation bias and selection on shaping codon usage patterns assuming a uniform mutation bias across the genome. However, mutation biases can vary both along and across chromosomes due to processes such as biased gene conversion, potentially obfuscating signals of translational selection. Moreover, estimates of variation in genomic mutation biases are often lacking for non-model organisms. Here, we combine an unsupervised learning method with a population genetics model of synonymous codon bias evolution to assess the impact of intragenomic variation in mutation bias on the strength and direction of natural selection on synonymous codon usage across 49 Saccharomycotina budding yeasts. We find that in the absence of a priori information, unsupervised learning approaches can be used to identify regions evolving under different mutation biases. We find that the impact of intragenomic variation in mutation bias varies widely, even among closely-related species. We show that the overall strength and direction of selection on codon usage can be underestimated by failing to account for intragenomic variation in mutation biases. Interestingly, genes falling into clusters identified by machine learning are also often physically clustered across chromosomes, consistent with processes such as biased gene conversion. Our results indicate the need for more nuanced models of sequence evolution that systematically incorporate the effects of variable mutation biases on codon frequencies.